The Long Game: Treatment, Management, and What Comes Next
Part Two of Two
This is Part Two of a two-part series on Cardiac Allograft Vasculopathy (CAV). Part One—“The Diagnosis Nobody Prepares You For”—covers what CAV is, the history of how medicine learned to detect it, and why the statistics that come up first on a search do not describe the situation most recipients are actually in. If you haven’t read it, start there.
The diagnosis has landed. The team has moved. Now the question is: what does managing this actually look like, day to day and year to year?
The answer is more multidimensional than most people expect. CAV management is not a single medication adjustment. It is a sustained, coordinated effort across immunosuppression, lipid pharmacology, metabolic management, diet, and surveillance—each component addressing a different part of the same underlying problem, most of them interacting with each other in ways that require ongoing calibration.
The Treatment Toolkit: More Than Medication
The Statin Question—and Why My Answer Changed
I want to be transparent about something, because it is directly relevant to how I think about my own treatment.
I have never been a proponent of statin therapy for the general population. My position has been, and remains, that the evidence for statins in primary prevention of cardiovascular disease in otherwise healthy people is considerably weaker than the medical establishment has historically represented—and that the risks of long-term statin use, including myopathy and diabetogenic effects at higher doses, are frequently underweighted against modest absolute risk reductions in low-risk individuals.
This is not that situation.
In heart transplant recipients, statins are doing something that goes well beyond lipid management. A major benefit appears to be immunomodulatory and pleiotropic—meaning the drug produces multiple beneficial effects through different biological pathways, including gene expression, simultaneously—not simply lowering cholesterol. That distinction is supported directly by the evidence.
The mechanism: statins in the presence of calcineurin inhibitors appear to reduce natural killer cell activity, suppress T-cell response, inhibit chemokine synthesis by mononuclear cells, and downregulate expression of MHC-II genes—the proteins that tag donor heart cells for immune recognition. They also upregulate endothelial nitric oxide synthase and downregulate growth factor genes responsible for smooth muscle cell proliferation. That smooth muscle proliferation is the direct mechanism of CAV. The landmark evidence came from Kobashigawa and colleagues in 1995 in the first randomized trial of pravastatin in heart transplant recipients, which found reduced CAV incidence, reduced rejection with hemodynamic compromise, improved survival, and—in a key subgroup—significantly reduced natural killer cell cytotoxicity. The first clinical signal that something immunological was happening alongside the lipid effect.
Strong evidence that lipid-lowering alone does not explain the statin benefit: a trial of PCSK9 inhibition—a class of injectable medications that lower LDL by 50% or more by blocking the protein that degrades LDL receptors in the liver—achieved profound LDL reduction in transplant recipients without changing intimal thickness at one year. This is documented in the 2025 University of Michigan review of CAV advances. If the statin benefit were driven primarily by lipid reduction, more aggressive lipid reduction should produce more benefit. It did not. The immunomodulatory and pleiotropic mechanisms appear to be doing the primary work.
This is why I take 30mg of rosuvastatin without reservation, despite my general position on statins. My team started me on pravastatin—the standard first-line choice, well-characterized immunologically from the Kobashigawa trial and with a safe interaction profile alongside tacrolimus. We moved through atorvastatin and eventually landed on rosuvastatin: high-intensity, with minimal CYP3A4 metabolism, which reduces the drug-level interference and myotoxicity risk that comes with atorvastatin or simvastatin when combined with tacrolimus and sirolimus. At 30mg, we are in high-intensity statin territory, indicated by both the CAV finding and elevated triglycerides at the one-year evaluation.
The Pharmacological Irony: Sirolimus and Triglycerides
Here is the complication that does not get enough discussion.
Sirolimus—the drug doing the most to slow CAV progression—is itself hypertriglyceridemic. The mechanism is specific: sirolimus inhibits catabolism of apolipoprotein B-100, raises circulating free fatty acid levels, and decreases lipoprotein lipase activity. Lipoprotein lipase is the enzyme responsible for clearing triglyceride particles from the bloodstream. When its activity is suppressed, triglycerides accumulate. Clinical data from patients switched to sirolimus shows an average 46% increase in serum triglycerides following conversion. If you pull my own bloodwork, you can see exactly the month sirolimus entered my regimen in the triglyceride trend line. The drug slowing the immune-mediated vascular disease is simultaneously worsening one of the nonimmune metabolic risk factors for that same disease.
This is not a reason to avoid sirolimus. The benefit substantially outweighs the liability. But it is a reason why triglyceride management becomes a specific and active concern from the moment sirolimus is started—and why the pharmacological approach to that management matters.
Vascepa: The Second Pharmacological Layer
In addition to rosuvastatin, I take icosapent ethyl (Vascepa) at 2 grams twice daily—the same dose used in theREDUCE-IT trial, published in the New England Journal of Medicine. REDUCE-IT enrolled 8,179 statin-treated patients with elevated triglycerides and demonstrated a 25% relative risk reduction for the composite cardiovascular endpoint—cardiovascular death, nonfatal MI, nonfatal stroke, coronary revascularization, or unstable angina. Icosapent ethyl is pharmaceutical-grade eicosapentaenoic acid (EPA), which reduces triglyceride synthesis in the liver and provides cardiovascular event reduction beyond what statins achieve alone.
Worth noting: other triglyceride-lowering therapies—niacin, fibrates—have not duplicated these cardiovascular outcomes. This points toward EPA having direct anti-inflammatory and plaque-stabilizing effects that are mechanistically independent of triglyceride reduction. A pattern consistently seen with statins: the drug is doing more than the simple lipid explanation accounts for.
Diet: What Actually Drives Triglycerides
Medication handles part of the triglyceride management picture. Diet handles the other part—and the mechanism matters enough to explain clearly, because the common intuition about what causes high triglycerides is frequently wrong.
Most people assume dietary fat drives triglycerides. The more accurate picture, particularly for patients with insulin resistance, is that refined carbohydrates and excess caloric intake are the primary lever—through a hepatic process called de novo lipogenesis.
The pathway works like this: when you consume carbohydrates—particularly refined carbohydrates and sugar—the resulting glucose and fructose are metabolized in the liver. When caloric intake from carbohydrates exceeds immediate energy needs, the liver converts the surplus into fatty acids, assembles those fatty acids into triglycerides, and packages them into VLDL particles for release into the bloodstream. Research is clearthat high-carbohydrate diets significantly increase this conversion process, while high-fat diets tend to suppress it. Dietary fat is not the primary lever—total caloric load and carbohydrate composition are.
For a transplant recipient on sirolimus—whose lipoprotein lipase activity is already pharmacologically suppressed, making it harder to clear triglyceride particles from the bloodstream—this matters considerably. The drug-induced impairment of triglyceride clearance compounds whatever dietary load enters the system. Reducing the refined carbohydrate load is not optional lifestyle management. It is a direct, mechanistic counter-intervention to the metabolic disruption sirolimus creates.
In practice: reduce refined carbohydrates and added sugar, prioritize whole food sources of energy, and pay attention to glycemic load rather than caloric totals alone. Alcohol is a secondary triglyceride driver and warrants reduction or elimination. Saturated fat affects LDL more than triglycerides specifically, though overall dietary quality remains relevant to the broader cardiovascular picture.
The Glycemic Connection
Triglyceride management and glycemic management are not separate concerns in a heart transplant recipient. They are deeply intertwined—and the drug regimen itself is partly responsible for the link.
Tacrolimus impairs insulin secretion by damaging pancreatic beta cells. Sirolimus causes peripheral insulin resistance. Running both—as most CAV-managed patients now do—creates a compounding diabetogenic effect from two different mechanisms simultaneously. Elevated blood glucose, in turn, drives increased hepatic conversion of excess glucose to triglycerides. The two numbers move together.
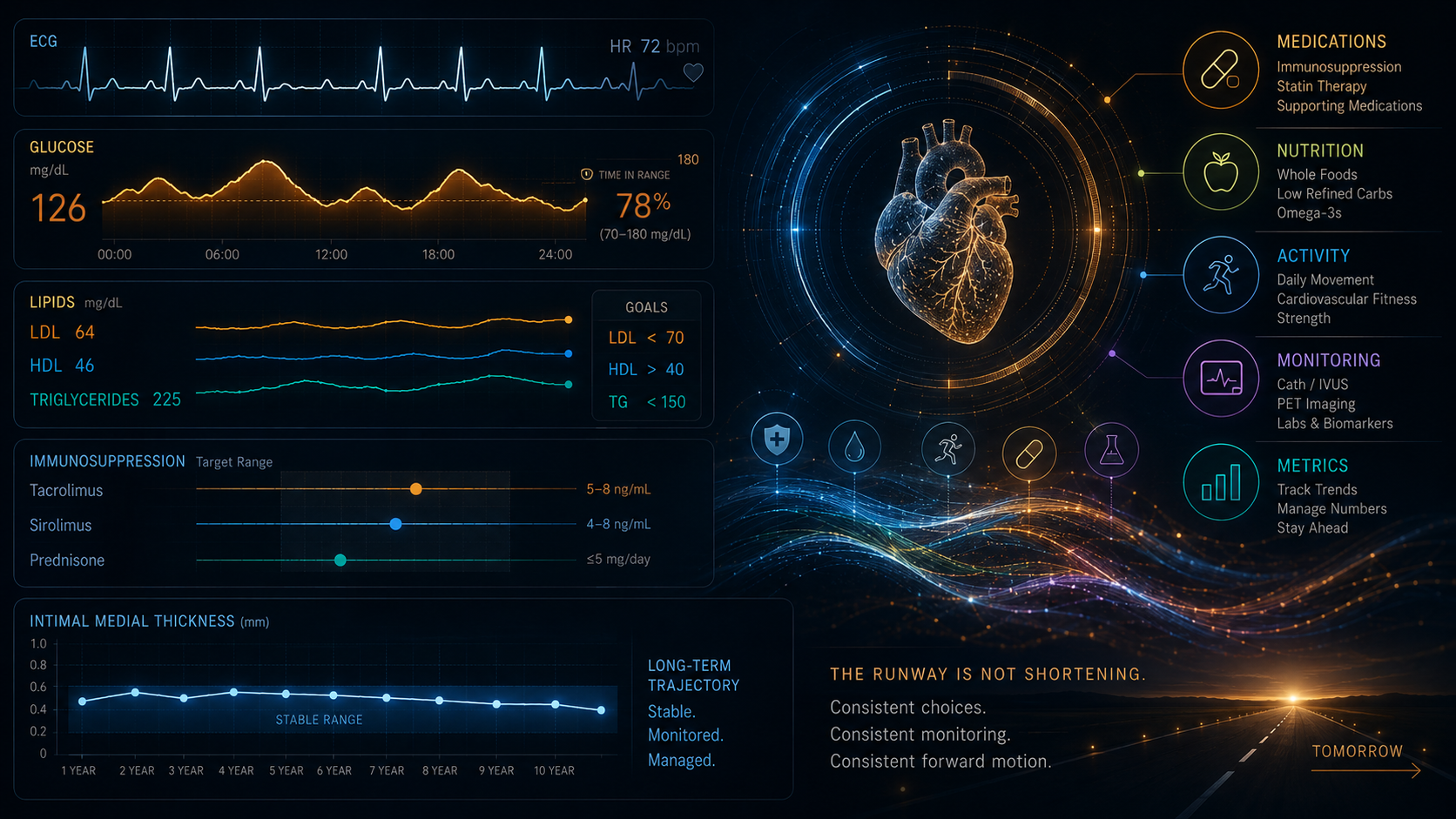
This is why glycemic management is load-bearing for overall metabolic risk in this population. The carbohydrate restriction that helps control triglycerides is the same intervention that moderates glucose excursions. Managing the metabolic consequences of immunosuppression is not a side issue—it is a direct component of CAV risk management. The SGLT2 inhibitor class—Jardiance (empagliflozin) specifically, which I take for glycemic control—adds another dimension worth noting. Beyond glucose management, the combined DAPA-HF and EMPEROR-Reduced meta-analysis demonstrated a 26% relative reduction in the combined risk of cardiovascular death or heart failure hospitalization, and a 25% reduction in recurrent heart failure hospitalizations or cardiovascular death—with consistent results in patients with and without diabetes. The cardiovascular protection is real and independent of the glycemic effect. In a CAV-managed patient whose primary long-term risk is cardiovascular, that is not a coincidental finding.
There is another dimension to the metabolic picture that rarely gets discussed because it sits at the intersection of pharmacology and lived experience. Tacrolimus and the broader immunosuppression regimen disrupt the ghrelin and leptin cycle—the hormonal feedback loop governing hunger and satiety. Ghrelin stimulates appetite and food intake; leptin suppresses it and promotes satiety. When this signaling is disrupted, the hypothalamic regulation of feeding breaks down. For many recipients, the result is persistent, pharmacologically-driven hunger that has nothing to do with caloric need—what those of us who experience it tend to call food noise. I was constantly hungry. Often famished. The predictable consequences were blood glucose swings and weight gain, both of which feed directly back into the metabolic risk picture for CAV. I approached my endocrinologist and suggested a GLP-1 receptor agonist. She agreed, and I was started on Rybelsus (semaglutide). GLP-1 receptor agonists decrease ghrelin levels and enhance leptin signaling, suppressing appetite through both central and peripheral mechanisms. I have reached the 14mg therapeutic dose. My weight is down more than 25 pounds. The food noise is all but gone. My A1c has dropped from 7.6 to 6.2, and fasting glucose is typically between 90 and 110. For a recipient managing CAV, those numbers are not incidental. They are load-bearing.
What Gets Watched and How
Managing CAV is not a passive exercise. The monitoring stack is specific and matters.
IVUS — remains the foundational gold standard for early, wall-based detection—tracking changes in vessel wall thickness that coronary angiography entirely misses. The finding that triggered my diagnosis was an IVUS measurement. Angiography that day showed nothing of concern.
Cardiac PET — has been incorporated into ISHLT guidance as an important noninvasive adjunct to invasive surveillance. It evaluates myocardial perfusion and metabolic inflammation, can detect microvascular disease that catheterization-based methods don’t capture, and reduces the procedural burden of monitoring over time. It has been added to my regimen going forward.
OCT — is now available at major transplant centers for cases requiring more detailed vessel wall characterization. It detected early-stage intimal thickening in 67% of vessel segments in one head-to-head study versus 14% by IVUS.
Lipid panel — runs at every visit. In a recipient with a CAV diagnosis, this is active management of modifiable nonimmune risk factors, not routine screening. Triglycerides are watched especially closely for recipients on sirolimus.
CK (creatine kinase) — is on the panel specifically because of the high-intensity statin. It is the early-warning system for myopathy—catching muscle stress before structural damage occurs.
Prospera — (donor-derived cell-free DNA) runs as the frontline noninvasive rejection screen. Subclinical rejection is a primary upstream driver of CAV; keeping rejection surveillance sharp is part of the CAV management picture.
What the Community Already Knows
The clinical literature tells one part of the story. The transplant community tells another.
When a post went up in a large heart transplant support group—a recipient six months out, asking whether CAV was something they could live with and treat, wondering what lifestyle changes might help—it drew more than ninety responses over the next few days. What came back was not a chorus of fear. It was something closer to institutional knowledge: accumulated across thousands of patient-years of lived experience, offered freely to a stranger who had just started worrying.
The range of that experience is the first thing worth noting. Jeffrey had his CAV caught at year one, made medication adjustments, and was writing from sixteen years out with no measurable increase. Eve was transplanted at thirteen and turned fifty-one the year she wrote her response, thirty-eight years post, with a recent echo showing no further changes. Mike included a photo of himself on a boat: “I am still living every day to the fullest.”
That is not a small data set. It is a human one, which is different and in some ways more useful.
When the Medications Work
Nearly every account of stabilized or improved early-stage CAV in the community threads involves the same inflection point: the team found it early, adjusted the medications, and things stopped moving.
Mike O.’s account is the most numerically specific. Diagnosed with severe CAV at his one-year evaluation—LAD artery 70% blocked—his team discontinued mycophenolate and added sirolimus. A follow-up catheterization three months later showed the blockage at 30%. His two-year evaluation: still 30%. His message to a newly diagnosed recipient:
“There is no such thing as a smooth heart transplant journey. Things are going to pop up and present themselves and your team is well prepared to manage them.”
And his bottom line on the drug that made the difference: “Sirolimus can do wonders.”
Josh had moderate CAV at year one. His team added sirolimus and adjusted his statin. His year-two IVUS showed no evidence of CAV. His message to the group was three words: “Hang in there.”
Others described the same pattern in varying degrees of detail: the switch to sirolimus or everolimus, the follow-up surveillance showing progression halted or reversed, the recalibration of the lipid panel and the slow settling into a new medication normal. Bill’s team found an issue at six months post, switched mycophenolate to sirolimus, and a year later the CAV was considered stable. His read on that outcome: “I can live with stable.” Jeffrey combined his sirolimus with dietary changes discussed with his team; at his next surveillance his CAV was measurably less than before. His team, he wrote, was amazed. Glenda set a goal of keeping her LDL under 70, added fish oil, and was seven years out with no progression and possibly slight improvement.
These accounts are not outliers. They are the modal experience of the community—which does not mean they are guaranteed, but they are what the evidence-based treatment protocol, applied early, tends to produce.
When It’s More Complicated
The community does not pretend otherwise about the cases where the front-line treatment didn’t work, or couldn’t be tolerated. Several members described working through multiple statins because of myopathy or other side effects before finding one that was tolerable. One member’s sirolimus experience included severe GI complications and hospitalizations serious enough that she ultimately refused the drug and told her team why; she eventually stabilized on an alternative regimen. Another stopped sirolimus after good early results because of kidney and GI effects and manages with diet and other interventions.
The community holds both realities—the transformative cases and the ones where the front-line drug had to be abandoned—without contradiction. The consistent message: the toolkit has more than one tool, and working through the options is part of the process, not a sign that the process is failing.
One detail from the threads that belongs in any honest discussion of the pharmacological picture: a member posted a clinical letter from a major academic transplant center informing them they had been randomized to the placebo arm in the PCSK9 inhibition after heart transplantation study—the same trial referenced in Part One showing that aggressive LDL reduction through PCSK9 inhibition did not improve intimal thickness or vessel function at one year. The human face that arrived with that finding is worth sitting with. The treatments that didn’t pan out are part of the story alongside the ones that did. The field is still learning.
The Long View
The person who has lived with CAV longest among the voices in these threads has been on her second heart for nearly four years after twenty-five years on her first. She serves as an administrator in the group and has watched hundreds of newly diagnosed recipients work through the same questions. Her framing of what CAV actually is deserves to be quoted directly:
“CAV is what I refer to as the game ender because inevitably, if you live long enough, it will cause your donor heart to fail. And that looms so incredibly large. But it varies so much. Sometimes progression is very slow—over decades. Sometimes so fast you barely have time to process it.”
She lived for decades waiting for the CAV-and-retransplant shoe to drop. She lived her life. When the time came, she went through it. She is still here, still moderating the group, still answering the questions of people who are—as she once was—reading scary numbers alone at night. Her advice on the anxiety itself: get support for it, because worrying, she said, is a huge complication in and of itself.
The retransplant accounts in the community are not uniformly grim. Kathleen was retransplanted after eleven years on her first heart and was fourteen years into her second when she wrote her response. Her message to someone newly facing that possibility: “It’s a lot easier. Your body is already used to the drugs, and I knew what to expect.” Gwen was diagnosed in 2010, converted to sirolimus which slowed progression significantly, eventually needed stents in 2022, and received a second transplant in late 2024—twelve years, she wrote, of it mostly not being an issue. A.J. was nine years out when she was retransplanted in late 2025 and at the time of writing was doing well.
There is also the surveillance anxiety that the community names without flinching—the texture of living between catheterizations, not just the moment of first diagnosis. Jason, two years out, had just completed his year-two left heart cath and was waiting for the cardiologist to call:
“I’m terrified of CAV progression. At year one I was CAV 0, with minimal findings under IVUS. I haven’t received my year-two results yet… one day at a time.”
That is where a significant portion of the CAV-diagnosed transplant population lives—not in crisis, but in the ongoing work of managing something that moves slowly and requires sustained attention. The community knows exactly where that person is, because most of them have been there.
What Year Two Actually Looks Like
I am writing this at eighteen months post-transplant. The day I finished this draft was the day of my eighteen-month clinical visit at The Christ Hospital in Cincinnati.
I discussed this piece with Dr. Sitaramesh Emani—my cardiologist at The Christ Hospital, and the kind of physician who engages substantively with a patient who shows up having done the research and wants to talk about it. His response was unambiguous: this kind of piece matters, because what patients find when they first search CAV is not representative of what a CAV diagnosis means at a center using current protocols. Both he and my transplant coordinator emphasized the same point: when IVUS surveillance turns up a subclinical signal, they move on it immediately. The threshold that triggered my diagnosis would have been below the clinical radar just a decade ago. Now we actually look for it. And now we know what to do when we find it.
That is not a small thing to say about a medical team. The Christ Hospital’s transplant program applies a conservative, patient-first approach that doesn’t wait for disease to declare itself before responding. They watch for everything. They jump on it early. They work with you rather than at you. In a field of medicine where the stakes are as high as they get, that culture of proactive, aggressive surveillance is not something to take for granted. It is something to seek out, and something to recognize when you have it.
The graft is running strong. The ejection fraction is solid. The pressures are where they should be. The biopsies have been clean throughout.
The CAV finding is real, and I am not minimizing it. It is a chronic condition requiring lifelong surveillance and metabolic discipline. I understand what poorly controlled CAV looks like at years five and ten, and that understanding shapes every decision I make today—about the medications, the diet, the glucose and triglyceride numbers, showing up for the caths and the PETs.
But the panic I felt that first night—the doom-scroll through statistics that were not describing my situation—that part I could have been spared with better information earlier.
If you’ve just received this diagnosis, you are reading this at the moment I was in. The numbers you’ve found online are real but they are not the whole picture. Your team caught it early, or they wouldn’t be talking about a subclinical measurement—they’d be showing you an angiogram with stenosis. They are moving on it, or you wouldn’t be reading about a medication change. The field has tools it didn’t have twenty years ago, and it has a treatment protocol that works. You are in the era those tools were built for.
Stephanie, diagnosed at three years out and writing at ten: “Don’t lose hope. Take your meds and live your life. We know it’s borrowed time as it is, so don’t waste it.”
The runway is not shortening.
Watch the numbers. Take the medications. Manage the carbohydrates. Show up for the caths and the PETs. And read the newer studies—not just the ones that come up first.
Key Terms
Full definitions at the One More Beat Glossary: onemorebeat.com/glossary/
De Novo Lipogenesis — The process by which the liver converts excess carbohydrates into triglycerides. The primary dietary driver of elevated triglycerides is carbohydrate intake, not dietary fat.
Pleiotropic — Producing multiple beneficial effects through different biological pathways, including gene expression, beyond a drug’s primary intended action.
PCSK9 Inhibitor — A class of injectable medications that lower LDL by 50% or more. A PCSK9 inhibition trial in transplant patients achieved significant LDL reduction without changing CAV progression—supporting the conclusion that statin benefits in this population are primarily immunomodulatory, not lipid-driven.
VLDL — Very Low-Density Lipoprotein. A particle the liver uses to transport triglycerides into the bloodstream. Elevated when de novo lipogenesis is high.
Ezetimibe (Zetia) — A cholesterol-lowering medication that reduces intestinal absorption of dietary cholesterol. Being considered as an additional lipid management tool in my regimen.
Further Reading
One More Beat Glossary: onemorebeat.com/glossary/
Kobashigawa et al. (1995), Pravastatin in Heart Transplant Recipients: PubMed
Stomberski & Colvin (2025), “CAV: Advances in Diagnosis and Management”: PubMed Central
Packer et al. (2020), EMPEROR-Reduced / DAPA-HF Meta-Analysis: The Lancet
Ghrelin/leptin mechanism: Collís et al. (2025), “The role of leptin and ghrelin in the regulation of appetite” — ScienceDirect
GLP-1 RA mechanism: Farooq et al. (2025), “Mechanisms of GLP-1 Receptor Agonist-Induced Weight Loss” — American Journal of Medicine
Bhatt et al. (2019), REDUCE-IT Trial: New England Journal of Medicine
ISHLT Registry Data: ishlt.org
Discover more from One More Beat
Subscribe to get the latest posts sent to your email.